Developing bioinformatic tools for detecting low-frequency genetic variants in large marine (meta)genomic data sets
Doctoral Researcher:
Marina Khachaturyan, GEOMAR - Helmholtz Centre for Ocean Research Kiel, mkhachaturyan@geomar.de
Supervisors:
-
Prof. Dr. Thorsten Reusch, GEOMAR - Helmholtz Centre for Ocean Research Kiel, Marine Evolutionary Ecology, treusch@geomar.de
-
Prof. Dr. Tal Dagan, University of Kiel, Institute for Microbiology, tdagan@ifam.uni-kiel.de
Location: Kiel
Disciplines: bioinformatics, genomics, metagenomics, population genetics, evolutionary biology
Keywords: mutation, re-sequencing, next-generation sequencing, adaptive dynamics, unequal read coverage, genetic elements
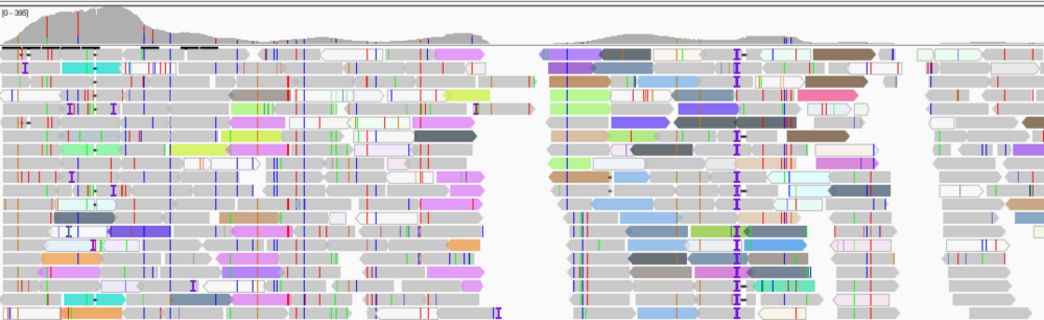
Background: Evolutionary novelty ultimately starts with singular events, a mutation (change of the primary DNA sequence) that by definition is exceedingly rare (1/population size). Mutations thus produce low-frequency variants that are difficult to detect in an ocean of common genetic variants. The same applies to novel microbial species that emerge via a lateral gene transfer, for example through the uptake of genetic elements such as plasmids. In order to understand evolutionary adaptive dynamics, including the nature and fitness spectrum of mutations and their temporal dynamics, the fate of such low-frequency variants is increasingly studied by ultra-deep DNA sequencing of genome and meta-genome (=mixed microbial) samples that yield billions of short sequence reads. The level of redundancy of DNA sequence reads, termed coverage, allows for the estimation of relative abundance of that genetic variant. Currently, there is a lack of systematic analytical assessments as to the repeatability, accuracy and potential bias of such variant detection at its lower detection limit. This problem is particularly pertinent as variable coverage within marine genomic and metagenomics samples is the norm when various genetic elements are pooled, e.g., (i) chromosomes and organelles or (ii) viruses, plasmids and their host.
Objective: The proposed project will assess in a systematic fashion the effects of unequal sequence coverage on the accuracy of genetic variant frequency estimation, with a special emphasis on rare mutations, microbes or genetic elements. Using statistically founded approaches, the dynamics of low-frequency variants in different examples of marine biological populations will be studied. In using different examples with unifying principles, this project aims to develop a conceptual framework for the comparative analysis of samples for low-variant detection and with unequal coverage. The project will address how different coverage introduces statistical biases in downstream analyses, and how rarity can be distinguished from the absence of particular genetic variants.
Aims: The specific data science aim will be to develop a robust comparative variant calling approach including a definition of confidence intervals and detection thresholds for the identification of genetic variants for different sequencing platforms with different error distributions, including the comparison of different sample groups for particular variant frequencies. The project will include the analysis of simulated data and analysis of actual marine high-throughput data. Marine examples where the novel tools will be applied to include (i) large asexually propagating organisms such as algae, corals and seagrasses (ii) plasmids or other genetic elements in metagenomics samples where we will compare the dynamics of low frequency elements having different copy number in an experimental evolution setup (iii) the rare ocean biosphere, i.e. low abundance taxa in metagenomic samples of microbes associated with marine host species where we explore statistical issues around the failure to detect certain variants that may be an important inference.
References
- Kupczok A, Neve H, Huang KD, Hoeppner MP, Heller KJ, Franz CAMP, Dagan T (2018) Rates of mutation and recombination in Siphoviridae phage genome evolution over three decades. Mol Biol Evol, 35, 1147–1159
- Olsen JL, Rouz. P, Verhelst B, ..., Reusch TBH, Van de Peer Y (2016) The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 530:331–335
- Wedemeyer, A., Kliemann, L., Srivastav, A., Schielke, C., Reusch, T.B., Rosenstiel, P., 2017. An improved filtering algorithm for big read datasets and its application to single-cell assembly. BMC Bioinformatics 18, 324